
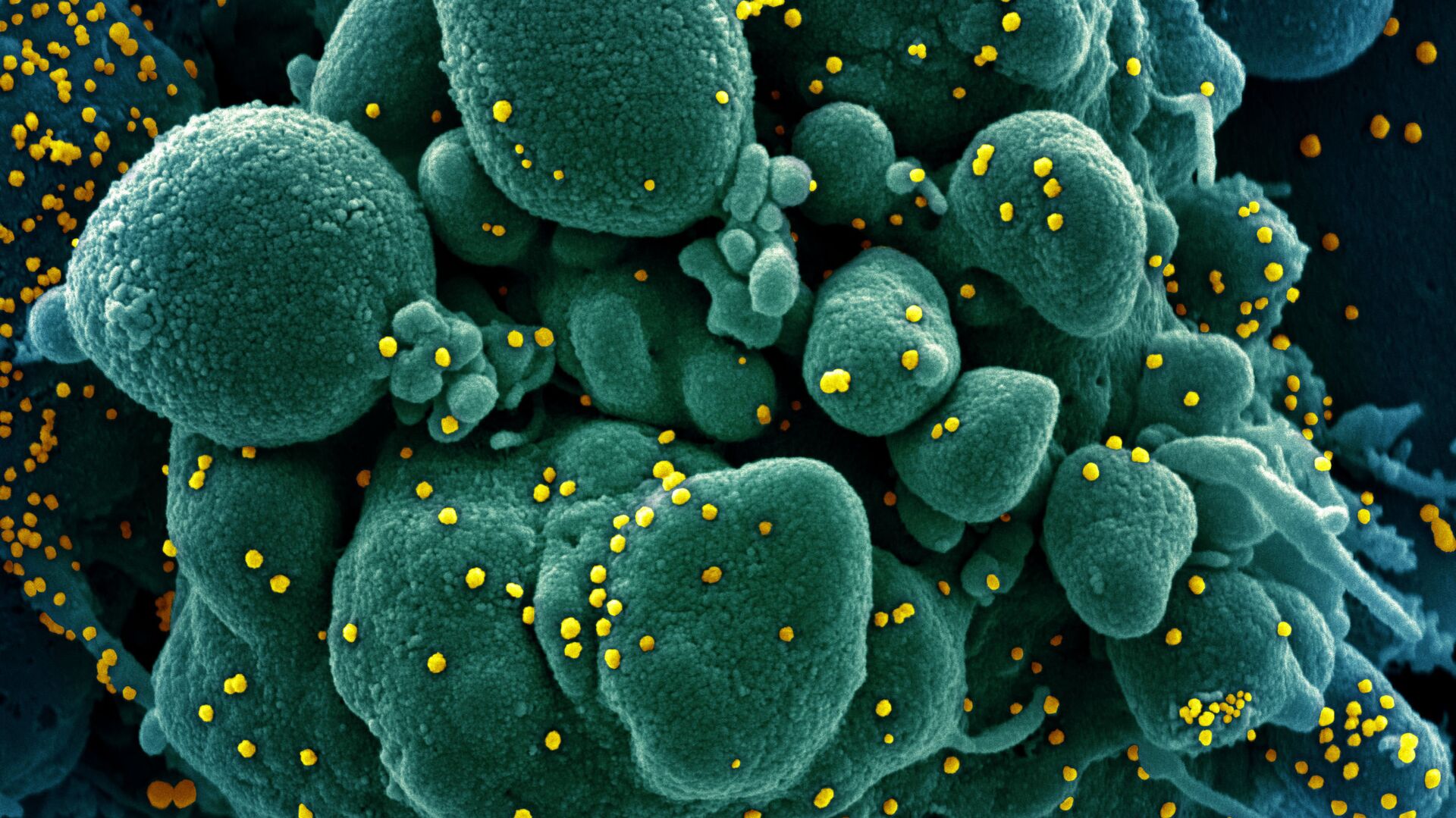
 infectada por el virus SARS-COV-2 (amarillo)")

Para el 17 de enero de 2020, cuando el virus aún no había sido importado desde Europa a México, el Instituto de Epidemiología (Indre) ya había desarrollado una prueba de PCR —reacción en cadena de la polimerasa— gracias a la información que compartieron los científicos chinos, antes de cumplirse el mes de haberse registrado el primer caso en la ciudad de Wuhan, en la provincia de Hubei.
"Nos preparamos para obtener la secuencia genética del virus en cuanto llegara su primera cepa al país", explicó el doctor Ernesto Ramírez, director del Instituto de Diagnóstico y Referencia Epidemiológica dentro de la Dirección General de Epidemiología federal mexicana.
Esto ocurrió el 27 de febrero de 2020 en la Ciudad de México, con un hombre que había estado durante dos semanas en Bérgamo, Italia, entre el 10 y el 12 de febrero de este año. Ramírez explicó que la muestra llegó al Indre a las 21:30 horas de ese día y antes de la medianoche ya se había confirmado la introducción del virus al país, con este primer caso.
Metagenómica
La cepa del primer caso importado a México confirmó su parentesco con las presentes en la región visitada por el paciente enfermo. Para el 1 de marzo, México había subido a la plataforma Gisaid la secuencia genómica de la cepa viral de su caso.
"Al igual que hicieron los científicos chinos, tenemos la obligación de compartir esta secuencia genómica en el Gisaid", explicó Ramírez.
El experto dijo que para identificar a la cepa se buscan asociaciones por medio de árboles filogenéticos.

Para identificar el flujo de importación del nuevo coronavirus a México, los científicos nacionales hicieron un análisis exhaustivo de los casos presentados durante los primeros quince días de ocurrencia de la pandemia en el país — entre el 27 de febrero y el 15 de marzo— período en el que colectaron muestras de carga viral suficiente para obtener de ellas las lecturas de una secuencia genómica completa en cada caso.
Durante los primeros 15 días de la pandemia en México, de 33 muestras que fueron tomadas de distintos pacientes se establecieron 17 secuencia del genoma de cada cepa.
Fueron incluidos pacientes con y sin antecedentes de viaje al exterior, pero sí con prueba confirmatoria de su infección con el nuevo coronavirus.
"Con eso fue suficiente para identificar que México tuvo dos vías de importación del nuevo coronavirus: por Europa y por Estados Unidos", informó el médico mexicano.
De las 17 cepas introducidas en el país que fueron identificadas durante la primera quincena de ocurrencia de casos en marzo del 2020, 15 de esas personas estuvieron primero en el Aeropuerto internacional de la Ciudad de México y de allí, se trasladaron a otros puntos de la Rrepública.
Estrategias de vigilancia genómica
El director explicó que a partir de este primer experimento de identificación de una serie de cepas en los primeros días cuando la pandemia no había alcanzado la dispersión comunitaria, se planificó una estrategia de seguimiento y vigilancia genómica de la presentación del nuevo coronavirus en México.

Este proyecto es ejecutado en conjunto con la Organización Mundial de la Salud y abarca el seguimiento de todos los tipos de virus activos, en particular, todos los coronavirus SARS-CoV-2. Esta clasificación se hace mediante dos sistemas: clados y linajes.
- En el mundo hay circulando seis clados del SARS-CoV-2y una serie más compleja y numerosa de linajes que "es más compleja porque la clasificación tiene que ver con los cambios en sus nucleótidos que implica el manejo de muchas letras para su nomenclatura".
- En México, de las 17 secuencias genómicas reconocidas a partir de los primeros quince días de pandemia, se identificó la presencia de cinco clados en 32 estados del país, de los seis en total que han sido reconocidos en el mundo.
- El clado con mayor presencia en México es el G, predominante en Europa, seguido en ocurrencia de los clados GR y GH. La presencia de los clados S y L, predominantes en Asia, sólo se registró en México durante el mes de marzo de 2020.
- Se identificaron 17 linajes del virus que son compartidos con los mayoritarios en el resto de América Latina: B.1; B.1.1; B.1.5 y B.1.5.11
"De la primera secuencia que pudimos compartir y obtener de estos genomas identificamos que el primer caso de México corresponde al clado GR, del linaje B.1", apuntó el científico.
"El proyecto de comorbilidades trabaja sobre 70 secuencias genómicas ya ensambladas en las que estamos analizando la severidad que presentó la enfermedad. Aunque a nivel internacional no se ha reportado ningún marcador molecular asociado con la severidad, pronto presentaremos las conclusiones de México al respecto", concluyó.


{kind=link}
{kind=link}
{kind=link}
{kind=link}